はじめに
様々な真核生物のゲノムには反復配列、リピート配列が存在します。
リピート配列は、それぞれの種あるいはその種を含むグループ(属、族、目)に特異的な配列も存在し、各生物種のゲノムに対してリピート配列をそれぞれ同定することが重要です。
RepeatModeler2( Flynn et al. 2020 )は反復配列の主要な構成要素である TE( 転移因子 transposable elements )をゲノム配列から検出し、そのライブラリーを作成するプログラムです。
ここではアズキ Vigna angularis の一品種の朱毬のゲノムを例に RepeatModeler2 の使用方法ならびに RepeatModeler2 で検出された配列を用いて RepeatMasker によりゲノム内での反復配列の場所を特定し、小文字に置換する(いわゆる soft-masking )方法を紹介します。
RepeatMasker によりゲノム内での反復配列の場所を特定し N 塩基として記載すること(いわゆる hard-masking )は RepeatMasker の出力オプションを設定することで実施できます。
(参考)
- Flynn et al. (2020) RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117:9451-9457. DOI:10.1073/pnas.1921046117
ゲノム配列の準備
まず公共データベース(米国国立衛生研究所)の NCBI( National Center for Biotechnology Information )に登録されているアズキ(朱毬)のドラフト・ゲノムの塩基配列をダウンロードします。
ここ をクリックするとデータを検索する画面が表示されます。
(2023年12月14日)
そこで、上のように赤で囲った枠から Genome を選択し赤矢印のように Vigna angularis を入力し Search をクリックすると、次のような結果が表示されます。
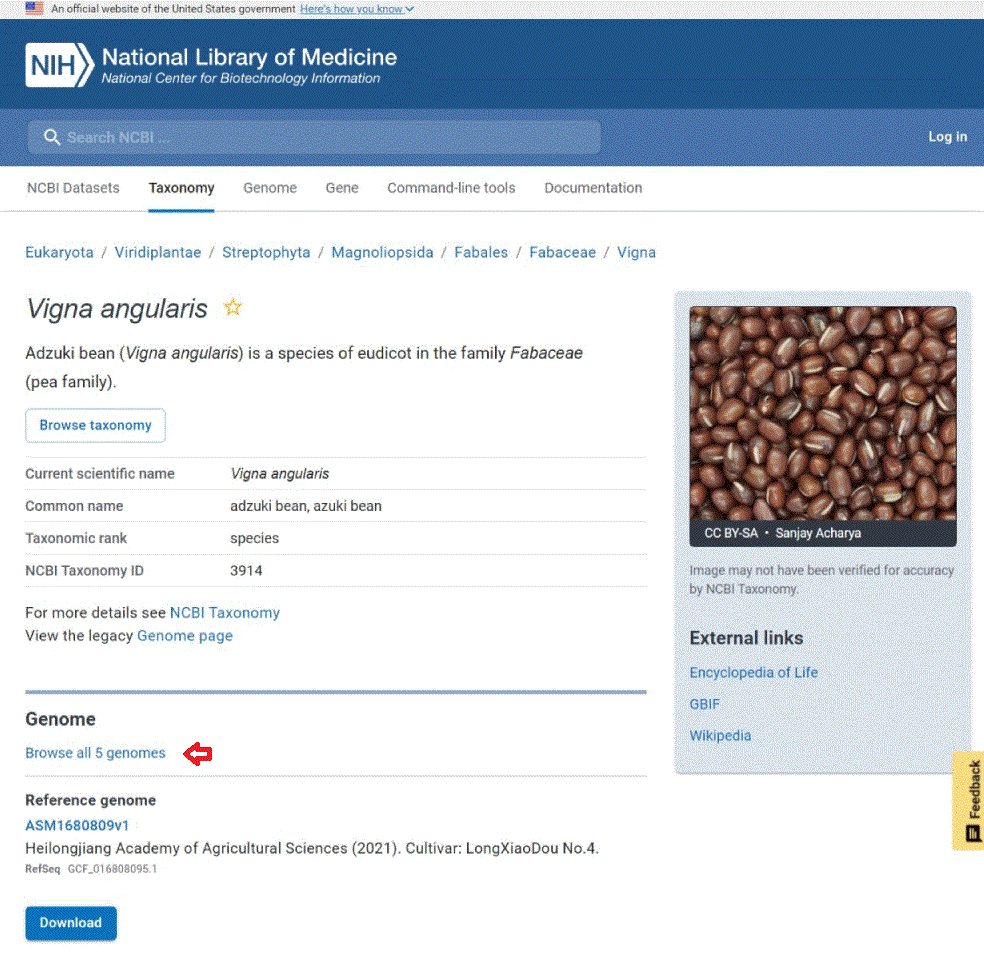
2023年12月14日時点での Reference genome は ASM1680809v1 で、 Heilongjiang Academy of Agricultural Sciences の研究グループによって 2021 年に登録された、品種 LongXiaoDou No.4 のゲノムです。ここの解析例ではそれより以前に完成された、朱毬のゲノム配列を使用します。 そこで同じ画面にある赤色の矢印の指す Browse all 5 genomes をクリックします。 すると
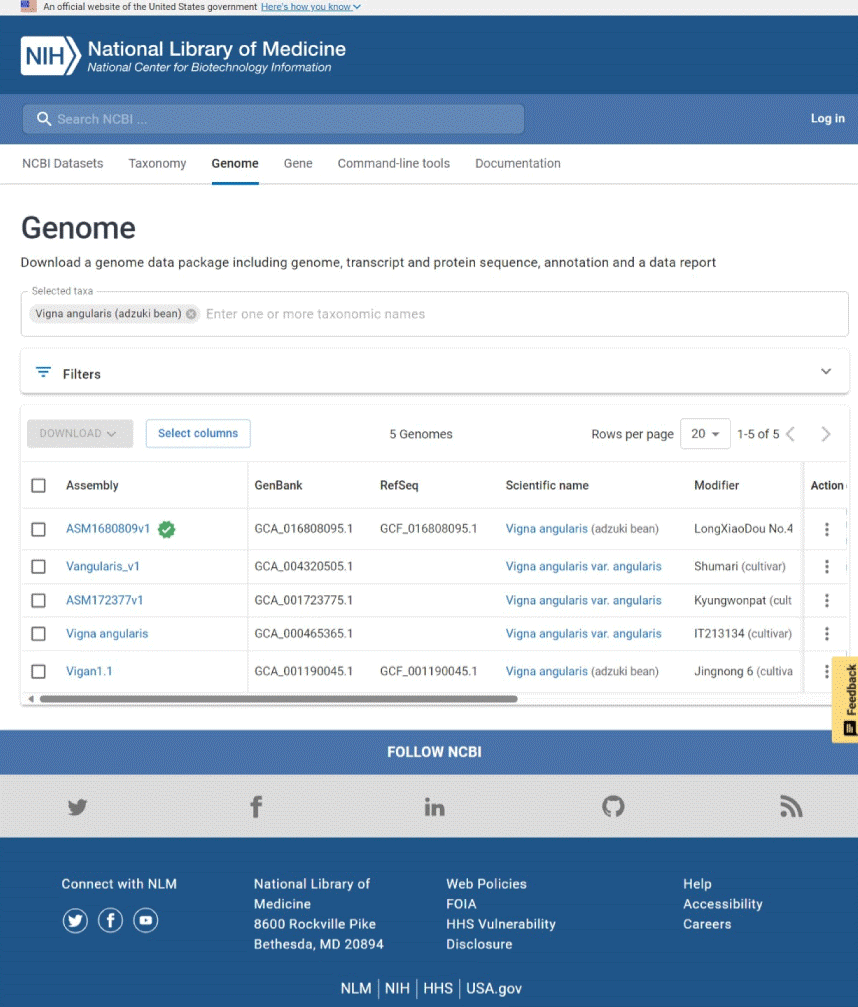
が表示されます。そこで朱毬( Shumari )のデータを含む Vangularis_v1 をクリックすると
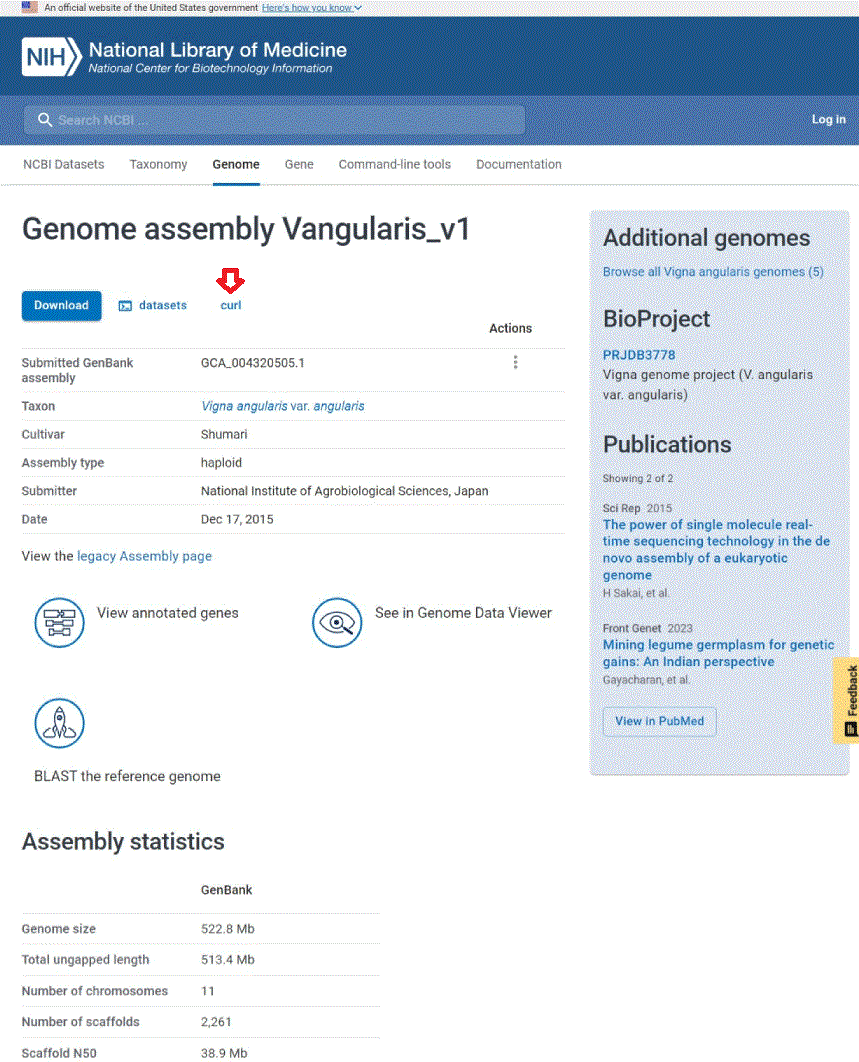
が表示されます。 このページでは朱毬のシークエンスの解析情報が詳細に記載されています。 赤矢印で指している curl をクリックしてポップアップして表示される Copy command をクリックすることで、ゲノム情報をダウンロードするための curl コマンドの引数を取得できます。そこで
curl -OJX GET "https://api.ncbi.nlm.nih.gov/datasets/v2alpha/genome/accession/GCA_004320505.1/download?include_annotation_type=GENOME_FASTA,GENOME_GFF,RNA_FASTA,CDS_FASTA,PROT_FASTA,SEQUENCE_REPORT"
のコマンドを実行し、ゲノムデータを圧縮された形式( ncbi_dataset.zip )としてダウンロードします。 次に
unzip ncbi_dataset.zip
とすると ncbi_dataset.zip ファイルが解凍されます。 ncbi_dataset 内の data ディレクトリには GCA_004320505.1 というディレクトリが存在し、このディレクトリに FASTA 形式で記載された GCA_004320505.1_Vangularis_v1_genomic.fna というゲノムの塩基配列のファイルが収められています。 このデータにいくつの配列があるかを調べるには、FASTA形式では各配列のタイトルが > で始まること(またその次の行から続く塩基配列には > がないことを)、すなわち > を含む行の数が配列数と一致することを利用し
grep ">" GCA_004320505.1_Vangularis_v1_genomic.fna | wc
あるいは
cat GCA_004320505.1_Vangularis_v1_genomic.fna | grep ">" | wc
とコマンドを実行することで、このファイルの中に 2261 個のシークエンスのタイトル、すなわち 2261 本の配列( scaffolds )が存在することが確認できます。 これは先の画面での The number of scaffolds の数と一致しています。
(参考)
RepeatModeler について
使用するプログラムである RepeatModeler の配布元は以下となります。(2023年12月14日時点)
| Program | RepeatModeler | |
| Latest version | RepeatModeler 2.0.5 (2023 OCt 5) | |
| Biocontainer | https://biocontainers.pro/tools/repeatmodeler | |
| Source | https://github.com/Dfam-consortium/RepeatModeler または http://www.repeatmasker.org/RepeatModeler/ |
ここでは現在国立遺伝学研究所・スーパーコンピュータシステムの Apptainer (旧 Singularity) コンテナに収納されている RepeatModeler を使用します。 現在利用可能なバージョンを次のコマンドで調べてみます。
ls -al /usr/local/biotools/r/repeatmodeler* /
すると、最新のバージョンは repeatmodeler:2.0.4--p15321hdfd78af_0となっています。 Biocontainer で既往解されている Latest version は 2.0.5 ですので最新のバージョンはまだ国立遺伝学研究所・スーパーコンピュータシステムにインストールされていませんでした。 もし最新のバージョンを利用したければ自らプログラムを配布元のサイトからダウンロードおよびインストールしてください。 インストールの手順も同サイトで公開されています。
repeatmodeler を用いた解析は次の3つの過程で実施します。
1.BuildDatabase によるデータベースの作成
2.RepeatModeler による反復配列のリストの作成
3.RepeatMasker によるマスキング
BuildDatabase によるデータベースの作成
まずは解析を行う作業用ディレクトリ(ここでは repeatmask とします)を作成します。
mkdir repeatmask
そして、作成したディレクトリに移動します。
cd repeatmask
次に、ドラフトゲノムの塩基配列の fna ファイルを Shumari.fasta としてコピーします。 下記で(パス名)には適切なパスを代入してください。
cp (パス名)/ncbi_dataset/data/GCA_004320505.1/GCA_004320505.1_Vangularis_v1_genomic.fna Shumari.fasta
データベース作成のためのスクリプト・ファイル Shumari.BuildDatabase.sh を次のように作成します。
#!/bin/sh
#$ -cwd
#$ -S /bin/sh
singularity exec /usr/local/biotools/r/repeatmodeler:2.0.4--pl5321hdfd78af_0 BuildDatabase -name "Shumari" Shumari.fasta
最後に Shumari.BuildDatabase.sh のジョブを投入します。
qsub Shumari.BuildDatabase.sh
これでデータベースの作成は完了です。参考までに朱毬・ドラフト・ゲノムの number of nucloetides は 522,761,097 bp で、プログラムの実行結果の概要は
| computer node | epyc | |
| running time | 12 sec | |
| maxvmem | 1.4G | |
| output files | Shumari.nhr Shumari.nin Shumari.njs Shumari.nnd Shumari.nni Shumari.nog Shumari.nsr |
となります。
RepeatModeler による反復配列のリストの作成
反復配列のリストの作成のためのスクリプト・ファイル Shumari.RepeatModeler.sh を次のように作成します。
#!/bin/sh
#$ -cwd
#$ -S /bin/sh
singularity exec /usr/local/biotools/r/repeatmodeler:2.0.4--pl5321hdfd78af_0 RepeatModeler -threads 12 -database Shumari -srand 20231220
ここで -database には先のプログラムで指定したデータベースの名前( Shumari )を引数として記載します。 また -threads 12 でスレッド数を、-srand で乱数発生の種( シード seed )の値を記載します。 -threads の数についてはジョブ投入時のオプション( -pe def_slot で指定する数 )とスーパーコンピュータシステムの空き状況を考慮して決定することになります。 乱数発生のためには必ずしもシードの値を記載する必要はありませんが、結果を再現できるようにするにはシードの値を定め記載しなければなりません(シードの値を記載しなければ、毎回異なるシードの値を用いてプログラムが実行されます)。 最後に Shumari.RepeatModeler.sh のジョブを投入します。
qsub -l d_rt=●●:●●:●● -l s_rt=●●:●●:●● -l medium -l s_vmem=〇〇G -l mem_req=〇〇G -pe def_slot 6 Shumari.RepeatModeler.sh
ここで -pe def_slot 6 としたのは同一計算ノード上の 6CPU コアを確保するためです。 medium を指定しているため Intel Xeon Gold 6148 (20 cores) Base 2.4GHz, Max 3.7GHz を搭載する Medium 計算ノードを使用します。 インテル® のHPで確認すると Intel Xeon Gold 6148 は 20 コア 40 スレッドの CPU ( 1 コアあたり 2 スレッド)であることから、 6CPU コアを確保することで 12 スレッドでの実行が可能となります(なると思っています)。 実行完了までの時間( ●●:●●:●● )と要求するメモリ量( 〇〇G )については予め把握することは簡単でありませんが、メモリ量を少なめに、また時間を少し長めにとって実行し(試行錯誤で)試してみてください。 参考までに朱毬・ドラフト・ゲノムの解析の場合は
| computer node | medium | |
| running time | 4 days 22 hours 0 min 30 sec | |
| maxvmem | 17.3G | |
| output files | Shumari-families.fa Shumari-families.stk Shumari-rmod.log |
となりました。実行中の過程・結果は RM_<PID>.<DATE> ディレクトリに記載されています。
(参考)
RepeatMasker によるマスキング
マスキングためのスクリプト・ファイル Shumari.RepeatMasker.sh を作成します。
#!/bin/sh
#$ -cwd
#$ -S /bin/sh
singularity exec /usr/local/biotools/r/repeatmodeler:2.0.4--pl5321hdfd78af_0 RepeatMasker -gff -xsmall -lib Shumari-families.fa Shumari.fasta
ここで -lib には RepeatModeler で出力されたファイルが引数となっています。 -xsmall を指定することでマスクする領域を小文字で記載( soft masking )します。 -gff を指定することでマスクする領域をリストした GFF( Gene Feature Finding )形式のファイルを追加出力します。 RepeatMasker には様々なオプション( hard masking を含む)が準備されていますので、各研究で引き続き行う解析の状況に応じてオプションを選択してください。 最後に Shumari.RepeatMasker.sh のジョブを投入します。
qsub -l s_vmem=〇G -l mem_req=〇G Shumari.RepeatMasker.sh
実際の朱毬のゲノムの解析の場合は、経験上(〇Gの代わりに)8Gのメモリを要求して実行しています。 このプログラムの実行結果は
| computer node | epyc | |
| running time | 6 hours 43 min 2 sec | |
| maxvmem | 6.6G | |
| output files | Shumari.fasta.cat.gz Shumari.fasta.masked Shumari.fasta.out Shumari.fasta.out.gff Shumari.fasta.tbl 等 |
となりました。 最終結果の概要は Shumari.fasta.tbl に記載されており、朱毬・ドラフト・ゲノムには
bases masked: 285152363 bp ( 54.55 %)
====================================================
number of length percentage
elements* occupied of sequence
----------------------------------------------------
Retroelements 90475 82388797 bp 15.76 %
SINEs: 472 134931 bp 0.03 %
Penelope: 119 33473 bp 0.01 %
LINEs: 3508 1076559 bp 0.21 %
CRE/SLACS 0 0 bp 0.00 %
L2/CR1/Rex 0 0 bp 0.00 %
R1/LOA/Jockey 0 0 bp 0.00 %
R2/R4/NeSL 0 0 bp 0.00 %
RTE/Bov-B 1042 190741 bp 0.04 %
L1/CIN4 2466 885818 bp 0.17 %
LTR elements: 86495 81177307 bp 15.53 %
BEL/Pao 2942 1305034 bp 0.25 %
Ty1/Copia 37254 27880796 bp 5.33 %
Gypsy/DIRS1 46299 51991477 bp 9.95 %
Retroviral 0 0 bp 0.00 %
DNA transposons 20896 15306954 bp 2.93 %
hobo-Activator 4055 1492603 bp 0.29 %
Tc1-IS630-Pogo 0 0 bp 0.00 %
En-Spm 0 0 bp 0.00 %
MULE-MuDR 3943 3129290 bp 0.60 %
PiggyBac 0 0 bp 0.00 %
Tourist/Harbinger 0 0 bp 0.00 %
Other (Mirage, 0 0 bp 0.00 %
P-element, Transib)
Rolling-circles 1401 657461 bp 0.13 %
Unclassified: 503016 157758116 bp 30.18 %
Total interspersed repeats: 255487340 bp 48.87 %
Small RNA: 874 1081860 bp 0.21 %
Satellites: 0 0 bp 0.00 %
Simple repeats: 134984 26344892 bp 5.04 %
Low complexity: 29686 1580810 bp 0.30 %
==================================================--
の反復配列があることが示されました。
解析結果のアーカイブ
ここまでの解析結果、すなわち作業用ディレクトリである repeatmask 内のファイルをまとめてアーカイブします。
cd ..
tar cjf Shumari.repeatmask.tar.bz2 repeatmask
最初の cd .. で作業用ディレクトリである repeatmask の親ディレクトリに移動します。 ディレクトリの関係を示すために、 ".." は親ディレクトリを、また "." は現在のディレクトリを意味します。 これらは相対的なディレクトリの位置関係を示すために非常に便利な記号です。
次の tar コマンドでディレクトリの内容を一つのファイルにまとめ、同時にファイルを圧縮します。 tar コマンドは元々磁気テープにファイルをまとめるコマンド( tape archives )でしたが、現在ではディレクトリ・ファイルのアーカイブをファイルとしてまとめ圧縮するコマンドとして広く利用されています。 ここでは、-c オプションで新規にアーカイブを作成、-j オプションで bzip2 形式に圧縮、-f オプションでテープの代わりに指定したファイルにアーカイブすることを記載しています。 これでこれまでの解析結果を、ディレクトリ構造やファイルの作成時間などを保ちつつ一括にダウンロードすることが比較的容易な作業となります。